あなたは、何百万人もの人々の健康と福祉に大きな影響を与えることができると信じている、新しく革新的な医療機器をお持ちだとします。しかし、その医療機器の市場を成功させるためには、まず、欧州連合(EU)、米国をはじめとする世界の主要な司法管轄区域における医療機器の審査・承認という、一見複雑なプロセスを乗り越えなければなりません。
また、医療機器の薬事承認を求めるのが初めての経験であれば、その道のりは確かに圧倒的に見えるかもしれません。
しかし、そのプロセスは見かけほど複雑ではありません。ここでは、ほとんどの主要市場で医療機器に適用される規制当局の審査・承認プロセスをガイドするための10ステップのロードマップを紹介します。
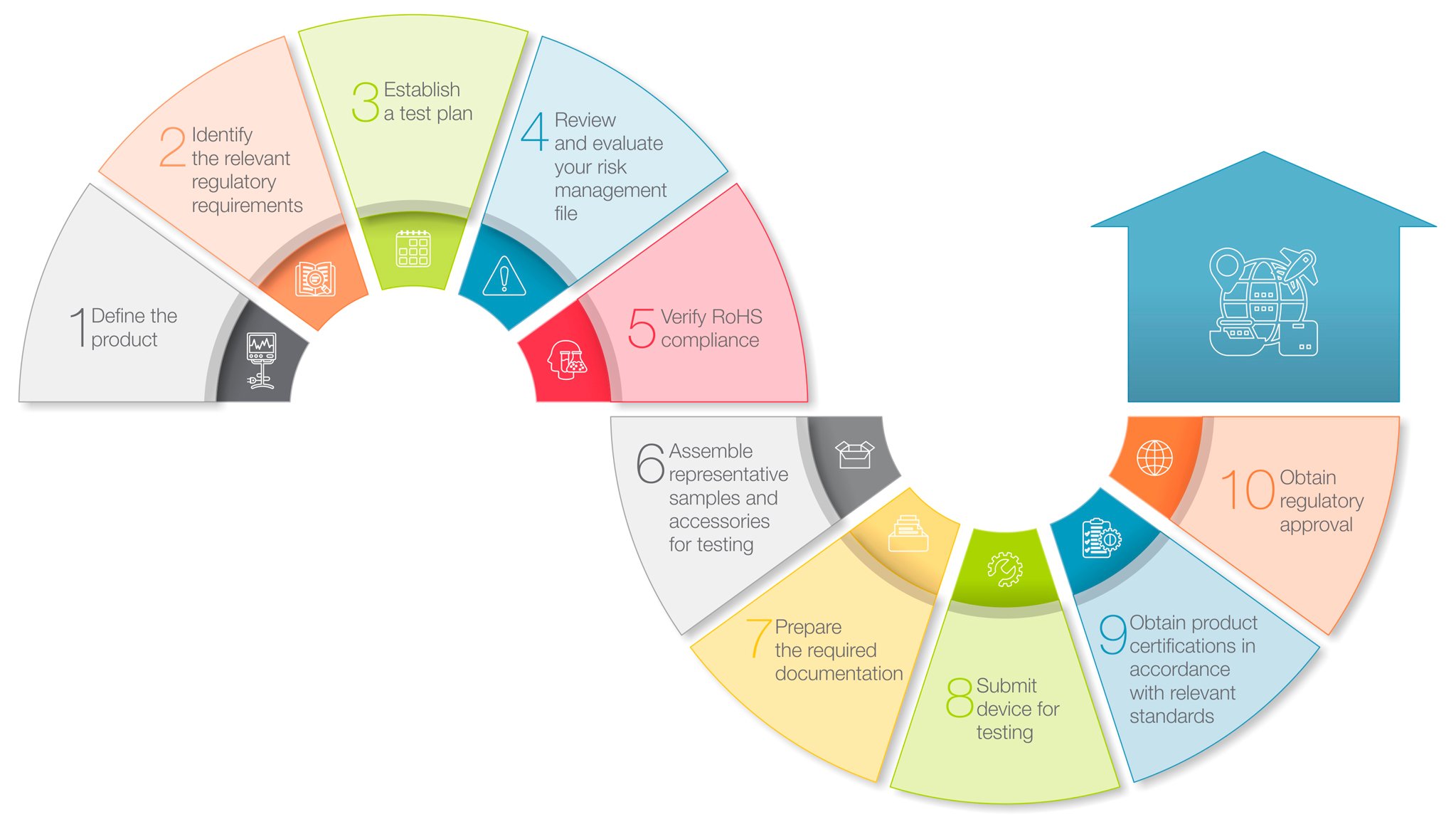
1. 製品の定義 
この最初のステップでは、製品がどこでどのように使用されることを意図しているかを確認します。例えば、確立された医療環境で訓練を受けた医療専門家によって使用される医療機器は、消費者や介護者による在宅医療環境での使用を意図した機器とは異なる要件が適用される場合があります。また、医療機器に適用される規制は国によって異なることが多いため、意図するターゲット市場を特定することも重要です。
2. 関連する規制要件を特定する 
ステップ1が完了したことで、医療機器の安全性と有効性を評価するためにどのような規制や規格が使用されるかを決定するために必要な情報が得られました。製品固有の要件に加え、多くの規制当局は、医療機器製造業者に対し、自社の品質管理システム(QMS)に関する文書の提出を要求し、ISO 13485のような適切なQMS規格の認証を要求する場合もあります。
3. 試験計画の策定 
御社の機器に適用される要件の詳細を理解した上で、御社の機器が関連する規制や規格に適合していることを証明するために必要なデータを作成する試験計画を策定しましょう。患者に対してより高いリスクをもたらす機器の場合、規制当局によっては、製品の安全性や有効性の主張を裏付ける証拠を作成するための臨床試験の実施も要求する場合があります。
4. リスクマネジメントファイルの見直しと評価
効果的なリスクマネジメント戦略は、医療機器の開発、製造及び市販後調査において重要な要素です。ISO14971の要求事項への適合は、所定の製品に関連するすべての潜在的な安全性リスクを特定し、評価し、リスクを許容可能なレベルまで低減するために必要なシステムと管理を実施するための効果的な枠組みを提供します。
5. RoHS適合の確認 
特定の管轄区域では、医療機器は有害とみなされる化学物質やその他の物質の使用制限の対象となっています。例えば、EUのRoHS(有害物質使用制限)指令では、鉛、水銀、カドミウム、フタル酸エステルなど10種類の禁止物質の医療機器への使用を明確に制限しており、さらに機器開発者に対して、関連要件への適合を証明する文書の作成を義務付けています。
6.試験用の代表的なサンプルとアクセサリーを組み立てる 
規制当局の審査及び承認プロセスにおける次のステップでは、試験の対象となる機器及び付属品の代表的なサンプルを選択し、組み立てます。サンプル機器と付属品は、最終製品の設計をできるだけ忠実に模倣し、想定される使用環境における機能性と安全性を徹底的に評価できるようにする必要があります。また、試験中に発生する可能性のある機器の故障や不具合を考慮し、十分な数のサンプルを準備してください。
7. 必要書類の準備
ステップ1~6までが完了したら、いよいよ試験所や規制当局にサンプルと一緒に提出する書類を準備しましょう。これには最低限、機器の技術ファイルまたは設計一式、臨床評価報告書、リスクマネジメントファイル、RoHS関連文書、すでに実施した試験の報告書などが含まれます。機器のリスク分類や市場特有の規制要件によっては、その他の文書が必要となる場合もあります。
8. 試験用デバイスの提出
医療機器の試験には、性能、機器の安全性、材料の生体適合性など、さまざまな側面が含まれます。また、機器の設計、特徴、機能によっては、放射・伝導エミッション、無線相互接続性、静電気放電(ESD)曝露に関連する潜在的リスクなど、電磁両立性(EMC)問題を評価することもあります。したがって、お客様のデバイスに必要なあらゆる試験機能を提供する試験所を選択することが重要です。
9. 関連規格に適合した製品認証の取得
試験結果を文書化した試験所発行の報告書は、医療機器が関連規格に適合し、製品認証を受ける資格があることを示す証拠となるはずです。場合によっては、貴社が発行する適合宣言書(DoC)などの自己認証で、特定の規制要件を満たすのに十分なこともあります。しかし、一部の規格については、EUノーティファイドボディのような認定された認証機関が試験データをレビューし、必要な認証を発行することを規制当局が要求する可能性があります。
10. 規制当局の承認取得
つまり、各規制当局による機器の承認が得られ、各市場において合法的に機器を販売することができるようになります。この最終審査・承認プロセスの厳しさやプロセスの詳細は、法域によって異なります。しかし、EUまたは米国で最初に規制当局の承認を取得することで、他の法域での承認プロセスを早めることができる場合が多いです。
すべての医療機器開発者と製造事業者が考慮しなければならない追加的な側面は、上記のすべてのステップを完了し、機器の規制承認を得るために必要な時間の長さです。
最初から最後まで、通常数ヶ月、場合によっては1年以上かかることもあります。
しかし、ここで紹介するロードマップに従うことで、あなたのデバイスが適用される要件を満たしているかどうかを早い段階で判断し、必要なデータと書類を確実に揃えることができ、試験や承認プロセスにおける驚きや予期せぬ遅れをなくすことができます。
詳細については、医療機器の規制要件についてご紹介するオンデマンド・ビデオをご覧いただくか、japan@nemko.com までお問い合わせください。
免責事項: 本記事は一般的な情報提供のみを目的としたものであり、コンサルティングや専門的なアドバイスを意味するものではありません。すべてのコンテンツは、掲載時点で知る限りにおいて正しいものですが、Nemkoは、提供された情報の正確性や完全性、なされた評価、または文書の使用から生じる結果について、いかなる責任も負いません。掲載内容に基づいて行動される場合は、事前にNemkoのサポートを受けてください。